HHT (Osler Weber Rendu)
 Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease and Osler–Weber–Rendu syndrome, is an autosomal dominant genetic disorder that leads to abnormal blood vessel formation in the skin, mucous membranes, and often in organs such as the lungs, liver, and brain.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease and Osler–Weber–Rendu syndrome, is an autosomal dominant genetic disorder that leads to abnormal blood vessel formation in the skin, mucous membranes, and often in organs such as the lungs, liver, and brain.
It may lead to nosebleeds, acute and chronic digestive tract bleeding, and various problems due to the involvement of other organs. Treatment focuses on reducing bleeding from blood vessel lesions, and sometimes surgery or other targeted interventions to remove arteriovenous malformations in organs. Chronic bleeding often requires iron supplements and sometimes blood transfusions. HHT is transmitted in an autosomal dominant fashion, and occurs in one in 5,000 people. It is lethal if you were homozygous for the disease.
The disease carries the names of Sir William Osler, Henri Jules Louis Marie Rendu, and Frederick Parkes Weber, who described it in the late 19th and early 20th centuries.[3]Osler Weber
Signs and symptoms
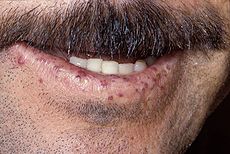
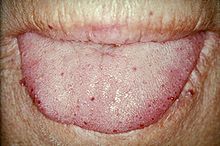 TelangiectasiasTongue telangiectases as seen in a person with hereditary hemorrhagic telangiectasia
TelangiectasiasTongue telangiectases as seen in a person with hereditary hemorrhagic telangiectasia
Telangiectasia (small vascular malformations) may occur in the skin and mucosal linings of the nose and gastrointestinal tract. The most common problem is nosebleeds (epistaxis), which happen frequently from childhood and affect about 90–95% of people with HHT. Lesions on the skin and in the mouth bleed less often but may be considered cosmetically displeasing; they affect about 80%. The skin lesions characteristically occur on the lips, the nose and the fingers, and on the skin of the face in sun-exposed areas. They appear suddenly, with the number increasing over time.About 20% are affected by symptomatic digestive tract lesions, although a higher percentage have lesions that do not cause symptoms. These lesions may bleed intermittently, which is rarely significant enough to be noticed (in the form of bloody vomiting or black stool), but can eventually lead to depletion of iron in the body, resulting in iron-deficiency anemia.
Arteriovenous malformations
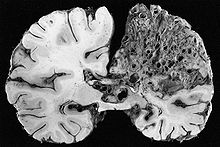 Arteriovenous malformation (AVM, larger vascular malformations) occur in larger org
Arteriovenous malformation (AVM, larger vascular malformations) occur in larger org
ans, predominantly the lungs (50%), liver (30–70%) and the brain (10%), with a very small proportion (<1%) having AVMs in the spinal cord.
Vascular malformations in the lungs may cause a number of problems. The lungs normally “filter out” bacteria and blood clots from the bloodstream; AVMs bypass the capillary network of the lungs and allow these to migrate to the brain, where bacteria may cause a brain abscess and blood clots may lead to stroke. HHT is the most common cause of lung AVMs: out of all people found to have lung AVMs, 70–80% are due to HHT. Bleeding from lung AVMs is relatively unusual, but may cause hemoptysis (coughing up blood) or hemothorax (blood accumulating in the chest cavity). Large vascular malformations in the lung allow oxygen-depleted blood from the right ventricle to bypass the alveoli, meaning that this blood does not have an opportunity to absorb fresh oxygen. This may lead to breathlessness. Large AVMs may lead to platypnea, difficulty in breathing that is more marked when sitting up compared to lying down; this probably reflects changes in blood flow associated with positioning. Very large AVMs cause a marked inability to absorb oxygen, which may be noted by cyanosis (bluish discoloration of the lips and skin), clubbing of the fingernails (often encountered in chronically low oxygen levels), and a humming noise over the affected part of the lung detectable by stethoscope.
The disesas is more prevelent in males with increased problems in females postmenapausally, thus estrogen has been proposed as a treatment. Reasonable to screen children of patients. Minimally with a CXR. Patients blood studies are completely normal. The anomaly is the absence of muscle, fibrin, and elastin in the walls of the blood vessels.
Nosebleeds
An acute nosebleed may be managed with a variety of measures, such as packing of the nasal cavity with absorbent swabs or gels. Removal of the packs after the bleeding may lead to reopening of the fragile vessels, and therefore lubricated atraumatic packing is recommended.[7] Some patients may wish to learn packing themselves to deal with nosebleeds without having to resort to medical help.[16]
Frequent nosebleeds can be prevented in part by keeping the nostrils moist, and by applying saline solution, estrogen-containing creams or tranexamic acid; these have few side effects and may have a small degree of benefit.[7] A number of additional modalities has been used to prevent recurrent bleeding if simple measures are unsuccessful. Medical therapies include oral tranexamic acid and estrogen; the evidence for these is relatively limited, and estrogen is poorly tolerated by men and possibly carries risks of cancer and heart disease in women past the menopause. Avastatin (an angiogenesis inhibitor) applie topically has been advocated. Nasal coagulation (with a deeply penetrating Argon laser) and cauterization may reduce the bleeding from telangiectasias, and is recommended before surgery is considered; often, several sessions are needed. It may be possible to embolize vascular lesions through interventional radiology; this requires passing a catheter through a large artery and locating the maxillary artery under X-ray guidance, followed by the injection into the vessel of particles that occlude the blood vessels. The benefit from the procedure tends to be short-lived, and it may be most appropriate in episodes of severe bleeding. Amicar, Septodermoplasty, medial maxillectomy have even been considered.
If other interventions have failed, several operations have been reported to provide benefit. One is septal dermoplasty or Saunders’ procedure,[17] in which skin is transplanted into the nostrils, and the other is Young’s procedure, in which the nostrils are meticulously sutured shut.
Experimental treatments
Several anti-angiogenesis drugs approved for other conditions, such as cancer, have been investigated in small clinical trials.[19] The anti-VEGF antibody bevacizumab, for instance, has been used off-label in several studies. In the largest study conducted so far, bevacizumab infusion was associated with a decrease in cardiac output and reduced duration and number of episodes of epistaxis in treated HHT patients. Thalidomide, another anti-angiogenesis drug, was also reported to have beneficial effects in HHT patients. Thalidomide treatment was found to induce vessel maturation in an experimental mouse model of HHT and to reduce the severity and frequency of nosebleeds in the majority of a small group of HHT patients. The blood hemoglobin levels of these treated patients rose as a result of reduced hemorrhage and enhanced blood vessel stabilization.
Epidemiology
The Netherlands Antilles, where HHT is more common than anywhere in the world, located off the coast of Venezuela.
Population studies from numerous areas in the world have shown that HHT occurs at roughly the same rate in almost all populations: somewhere around 1 in 5000. In some areas, it is much more common; for instance, in the French region of Haut Jura the rate is 1:2351 – twice as common as in other populations. This has been attributed to a founder effect, in which a population descending from a small number of ancestors has a high rate of a particular genetic trait because one of these ancestors harbored this trait.[8] In Haut Jura, this has been shown to be the result of a particular ACVRL1 mutation (named c.1112dupG or c.1112_1113insG).[2] The highest rate of HHT is 1:1331, reported in Bonaire and Curaçao, two islands in the Caribbean belonging to the Netherlands Antilles.
Most people with HHT have a normal lifespan.[1] The skin lesions and nosebleeds tend to develop during childhood. AVMs are probably present from birth, but don’t necessarily cause any symptoms. Frequent nosebleeds are the most common symptom and can significantly affect quality of life.[7]
History
Several 19th century English physicians, starting with Henry Gawen Sutton (1836–1891)[23] and followed by Benjamin Guy Babington (1794–1866)[24] and John Wickham Legg (1843–1921),[25] described the most common features of HHT, particularly the recurrent nosebleeds and the hereditary nature of the disease. The French physician Henri Jules Louis Marie Rendu (1844–1902) observed the skin and mucosal lesions, and distinguished the condition from hemophilia.[26] The Canadian-born Sir William Osler (1849–1919), then at Johns Hopkins Hospital and later at Oxford University, made further contributions with a 1901 report in which he described characteristic lesions in the digestive tract.[27] The English physician Frederick Parkes Weber (1863–1962) reported further on the condition in 1907 with a series of cases.[28] The term “hereditary hemorrhagic telangiectasia” was first used by the American physician Frederic M. Hanes (1883–1946) in a 1909 article on the condition.
The diagnosis of HHT remained a clinical one until the genetic defects that cause HHT were identified by a research group at Duke University Medical Center, in 1994 and 1996 respectively.[9][10] In 2000, the international scientific advisory committee of HHT Foundation International published the now widely used Curaçao criteria.[7][15] In 2006, a group of international experts met in Canada and formulated an evidence-based guideline, sponsored by the HHT Foundation International.